
【鄭智文藥師】漫漫新藥路(下)
什麼是解盲?
我們常常從新聞中聽到某一新藥進行「解盲」,這是因為藥物試驗會用到雙盲試驗,即病人被隨機編入對照組與實驗組,對照組給予安慰劑,實驗組給予真正某藥物,所以病人與研究觀察人員都不知道誰得到真正的藥物,直到研究結束才進行資料分析,然而大規模的試驗會設計期中分析(interim analysis)以幫助瞭解未來整個試驗的成功率與可能風險。解盲結果如果達到統計學上的顯著意義(p值<0.05),簡單解釋就是有100人進行雙盲試驗,有95人有達到預期效果,則為解盲成功;若是解盲失敗(p值>0.05)也不是完全失敗,畢竟臨床解盲數據非常複雜,除非療效極好或極差,不然絕大部分藥物試驗結果都落在中間值,研究人員必須就存活期、病人反應、生活品質等多項分析,以瞭解對哪些族群是有效的,以及後續是否有商業開發價值等條件評估,再重新設計條件較有利的臨床試驗,以取得臨床成功的可能。
新藥查驗及上市
在所有三個階段的臨床試驗成功完成後,可向衛生主關機關提交新藥查驗登記(New Drug Application, NDA)申請藥證,製藥公司必須能夠清楚地證明藥物的安全性與有效性,並提供藥物在臨床試驗中收集的所有科學信息(製造與管理、毒理與藥理、藥動與藥效、臨床試驗)。一般來說,新藥查驗可能需要至少6個月的時間審查,根據IRPMA調查報告,若是創新藥物在美國新藥查驗的總時間為390天,而台灣需要668天,創新藥物審查通過後取得專利藥物許可證,能享有5年資料專屬期(Data exclusivity)的保護,學名藥不得引用原廠藥的試驗資料作為申請上市的依據,各國對資料專屬期的時間長度的規定各有差異。
另外,某新藥已經在某一地區證實其有效性與安全性而且上市之後,這種新藥若是要在另一個地區註冊上市,而新藥在新地區執行有效性與安全性的臨床試驗稱作銜接性試驗(Bridging Study Evaluation, BSE),近年來亞洲與歐美同步進行臨床試驗的案件越來越多,這樣可加速藥物發展與縮短上市許可的時間,讓新藥可以同步申請許可,以及同步通過上市,以避免延遲病人使用新藥的契機。
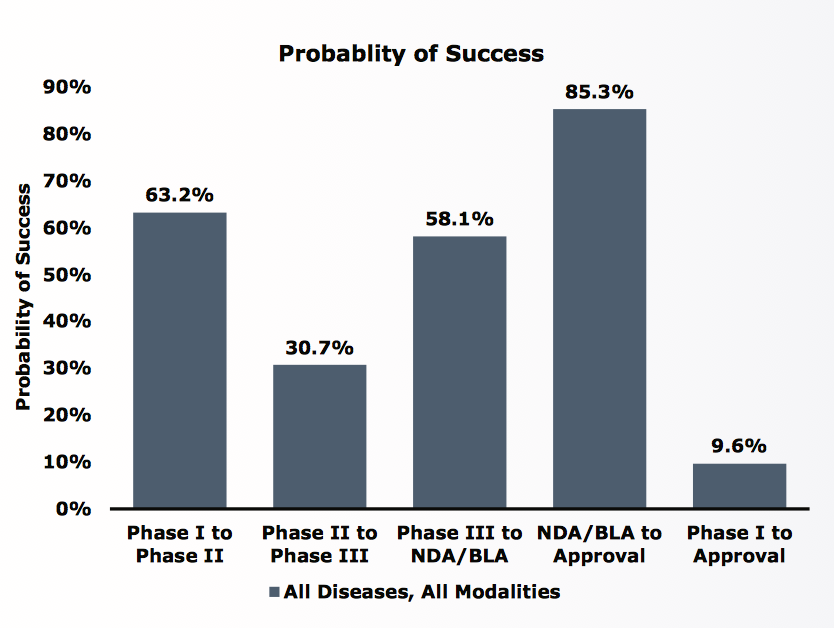
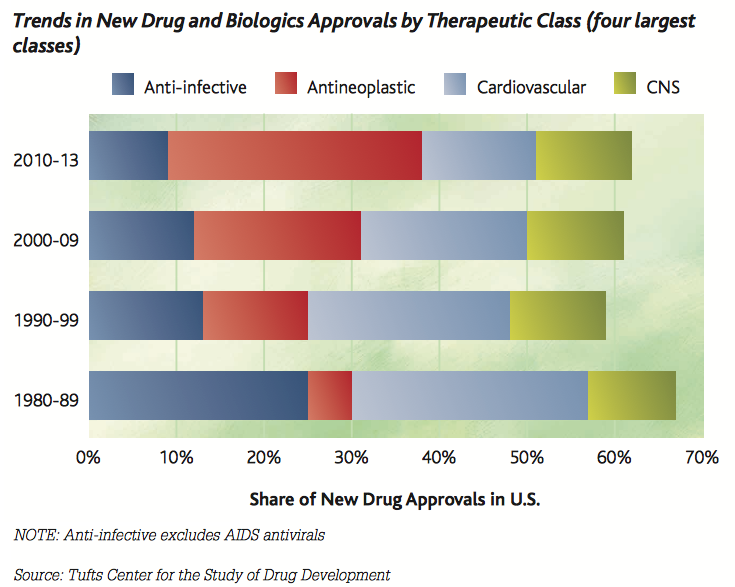
根據Biomedtracker資料,針對2006至2015年這十年間,全球1103家公司研發中新藥9985件來探討,臨床試驗第一期到新藥審查核准上市的總體成功率為9.6%。新藥自臨床第一期進入第二期的成功率為63.2%,自第二期進入第三期成功率為30.7%,自第三期完成到新藥審查成功率58.1%。現在新藥的研發主要有四大類趨勢:抗感染藥物(anti-infective)、抗腫瘤藥物(antineoplastic)、心血管藥物(Cardiovascular)與中樞神經藥物(Central Nervous System, CNS),早期著重於抗感染藥物與心血管藥物的研發,近十年來重心轉向抗腫瘤藥物,而神經精神藥物的研發一直是關注重點。
新藥取得藥證上市後,進入臨床第四期,雖然藥品上市前所做的臨床試驗已經提供充分的療效證據,但往往因為試驗設計與排除條件的限制,侷限了藥品安全性的資料。依循藥品優良安全監視規範(Guidance for Good Pharmacovigilance Practice),新藥自發證日起五年為藥品安全監視期,目的是減少藥物危害的風險。當藥物發生基於證據、或是可能因果關係,判定在任何劑量下對藥品所產生有害的、非蓄意的個體反應(即藥物不良反應),可由醫療機構、藥廠或民眾向全國藥物不良反應通報中心來通報或查詢,確保用藥安全。
新藥研發是必須集合人才、技術、資金、智慧財產、臨床試驗等各項高知識密集的產業鏈,最終佈局在全球市場,可以說是高投資、高風險但獲利可觀
根據GEN的報告,2016年銷售冠軍的Humira(復邁,用於自體免疫疾病)其年度銷售總額為160億美元,前十名的銷售總額最少也有57億美元。台灣三十多年來致力於生技醫藥產業之發展,一步一步穩紮穩打,終於在2014年由TFDA審查通過第一個由國人研發的創新成分藥物-Nemonoxacin太捷信®,為台灣新藥研發樹立重要里程碑,接著若能拿到歐美藥證打入國際,即可向世界證明台灣優異的研發能力,台灣生技醫藥在國際發光發熱,指日可待。

杏隆藥局 諮詢藥師
問8 線上視訊諮詢
諮詢專業醫師的第二意見,確保您得到最適合的治療方案
您可能想知道




追蹤問8粉絲專頁